

About Huntington Disease
Huntington Disease is an inherited disorder commonly referred to as HD. It’s caused by a defective gene containing a tri-nucleotide repeat (CAG) on chromosome four.
Genetic markers were discovered in 1983 leading to a breakthrough a decade later when scientists finally located the defective gene responsible for Huntington Disease. This allowed for the first time an accurate test to be developed.
This type of “triplet” gene causes nerve cells in the brain to deteriorate, producing symptoms difficult to predict in either their arrival or their course.
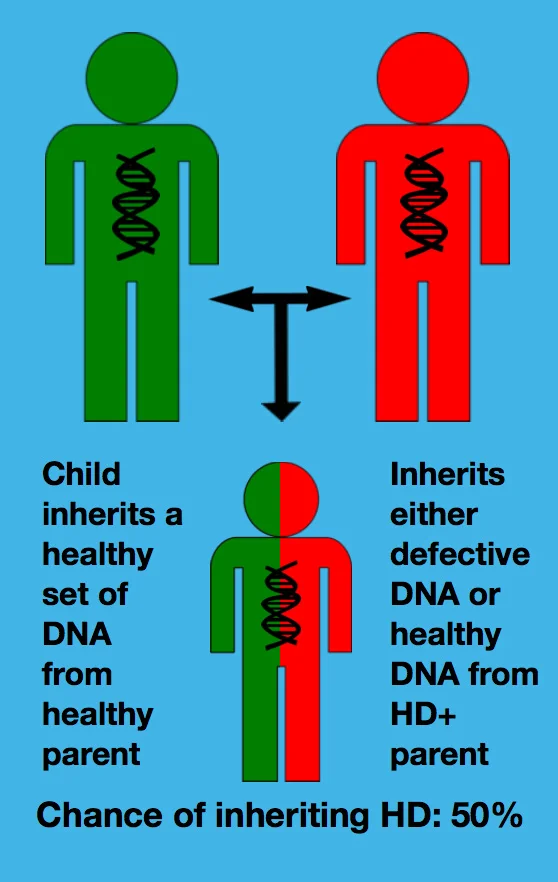
Every child of a person with Huntington Disease has a 50% chance of inheriting the defective gene and developing symptoms. It depends solely on which chromosome four is passed down from the HD affected parent.
Huntington Disease causes brains cells to deteriorate and die at a rapid pace. Symptoms generally begin between the ages of 30 and 50 but can strike at any age.
As more and more brain cells disappear, the functions they controlled will begin to fail.
The cellular damage caused by Huntington Disease is permanent and irreversible.
The History of Huntington's
While references to diseases involving involuntary movement or chorea (dancing mania) date back to the Middle Ages, the first mention of the symptom being caused by a hereditary disorder didn’t occur until the 1800s.
Huntington described the affliction “hereditary chorea,” notable for it’s hereditary nature, a tendency to dementia, and it’s manifesting itself as a grave disease only in adult life.
The disease had been poorly understood through the 18th and 19th centuries because many with the genetic defect died before onset of symptoms. As life spans expanded, symptoms had more time to manifest and more carriers were discovered.

Dr. George Huntington is credited with discovering the disease after writing an article for a medical journal based on his studies of several generations of afflicted families living in Long Island, N.Y. in 1872.